


Hirayama disease: Introduction
Hirayama disease is a form of motor neuron disease characterized by weakness & wasting of distal upper extremity effecting C5 to T1 level with predominant involvement of C7-T1 segmental myotomes 1, 2. It is a non-progressive disease which is insidious in onset with male predominance. The disease was first described by Keizo Hirayama in 19513. There is sparing of the brachioradialis muscle producing characteristic oblique amyotrophy. The condition is commonly unilateral, may be asymmetrically bilateral & rarely symmetrical. Rarely there may be involvement of the lower limbs.
Pathophysiology
The disease is thought to be due to discordant growth of the vertebral column & spinal canal contents resulting in tight dural sac. In normal spine the dura is attached to foramen magnum, C2 & C3 cranially and coccyx caudally. In between, the dura is also anchored to the vertebral canal at the exit of the nerve roots4. There is atleast a discrepancy of 5 cm in the change in length along the anterior & posterior surface of the cervical canal in flexion5. In normal subject, there are several transverse folds which compensate for the increased length of the cervical canal in flexion, however in Hirayama disease there is lack of these dural folds resulting in dural stress5. As a result, the dura is displaced anteriorly with resultant compression of the spinal cord. The disproportionate shortening of the dural sac is accentuated during the juvenile growth spurt6. Flexion of the neck causes loss of posterior dural attachment. Dural sac is displaced anteriorly & causes dynamic narrowing of the canal resulting in compression of cervical cord predominant at C7-T1 level7, 8. Chronic repeated compression of the cord causes microcirculatory changes in the region involving the territory of anterior spinal artery. This results atrophy of the cord predominantly involving the anterior horn as white matter is resistant to ischaemia9.
Imaging
Clinically these patients present with insidious onset of muscle weakness & atrophy in the hand & forearm with sparing of the brachioradialis. Changes are generally unilateral or asymmetrically bilateral & rarely symmetrically bilaterally.
On static MR imaging, there is focal cord atrophy, asymmetrical cord flattening, T2 hyperintensity in the cord & loss of attachment of the dura with lamina. Dynamic flexion study shows separation & anterior displacement of posterior dural sac, widening of lamino-dural space, multiple flow void signals due to venous engorgement and post contrast study showing enhancing component in the epidural space.
Differential diagnosis includes syringomyelia, cervical spondylosis, spinal cord tumor, amytrophic lateral sclerosis & traumatic myelopathy. These conditions can be differentiated by proper clinical history & imaging features.
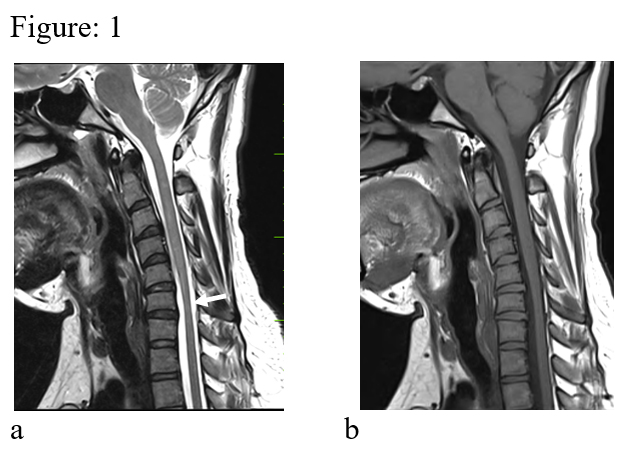
Figure: 1 (a & b). Sagittal T2 & T1 images in neutral position reveal localized cord atrophy (white arrow). No demonstrable dural displacement is seen.
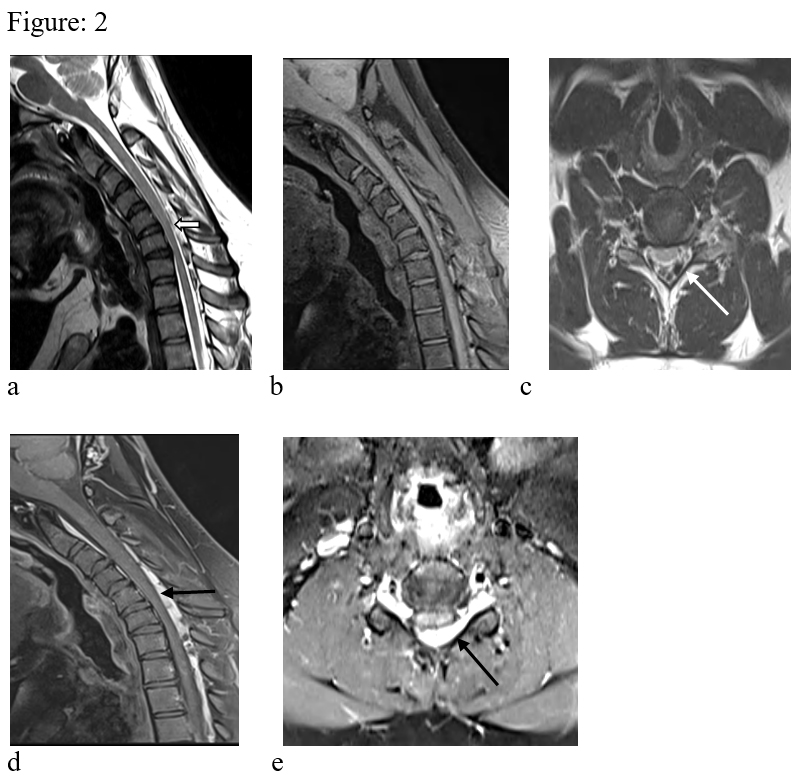
Figure: 2. T1 & T2 weighted images in flexion in sagittal (a, b) & axial (c) reveals anterior displacement of the dura (white block arrow) causing compression of the cervical cord. There is widening of the spino-dural space with prominent epidural veins (white arrow). The cervical cord shows atrophy. Sagittal & axial post contrast images in flexion (d, e) shows thick heterogeneously enhancing component in the epidural space (black arrow). There is also extension of the enhancing epidural lesion to the upper dosral region.
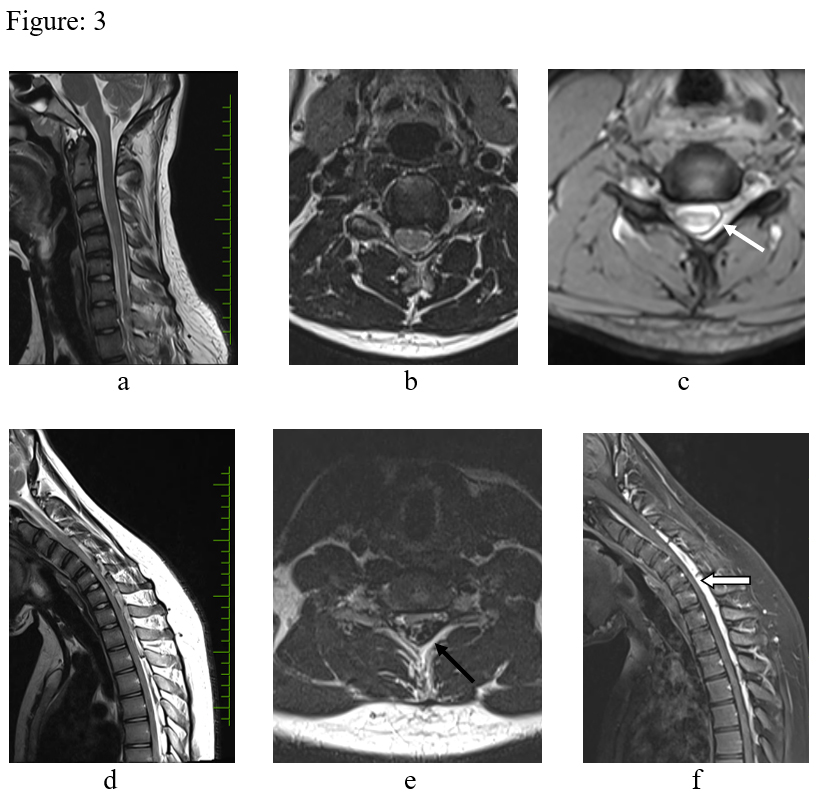
Figure: 3 – T2 sagittal (a) T2 axial (b), medic axial (c) in neutral position. There is focal cord atrophy, asymmetrical cord flattening & loss of attachment of the dura with lamina (white arrow). d, e & f – Flexion study (d, e – T2 sagittal & axial, f post contrast T2 sagittal) – There is anterior displacement of the dural sac, widened lamino-dural space with venous engorgement (black arrow) and epidural enhancing component (block arrow).
References:
- Hirayama K, Toyokura Y, Tsubaki T. Juvenile muscular atrophy unilateral upper extremity a new clinical entity. Psychiatr Neurol Jpn. 1959;61:2190–2197.
- De Carvalho M, Swash M. Monomelic neurogenic syndromes: a prospective study. J Neurol Sci. 2007;263:26–34. [PubMed] [Google Scholar]
- Hirayama K, Toyokura Y, Tsubaki T. Juvenile muscular atrophy of unilateral upper extremity; a new clinical entity. Psychiatr Neurol Jpn. 1959;61:2190–7.
- Monali Raval, Rima Kumari, Aldrin Anthony Dung Dung, Bhuvnesh Guglani, Nitij Gupta, Rohit Gupta MRI findings in Hirayama disease. Indian J Radiology Imaging.2010 Nov; 20(4): 245–249.
- Huang Y-L, Chen C-J. Hirayama disease. Neuroimaging Clin N Am. United States; 2011 Nov;21(4):939–50, ix – x. (PMID: 22032508)
- Mukai E, Sobue I, Muto T, Takahashi A, Guto S. Abnormal radiological findings on juvenile-type distal and segmental muscular atrophy of upper extremities. Clin Neurol. 1985;25:620–6. [PubMed] [Google Scholar]
- Hirayama K. Juvenile muscular atrophy of distal upper extremity (Hirayama disease). Intern Med. JAPAN; 2000 Apr;39(4):283–90. (PMID: 31011327)
- [4] Hassan KM, Sahni H. Nosology of juvenile muscular atrophy of distal upper extremity: from monomelic amyotrophy to Hirayama disease–Indian perspective. Biomed Res Int. United States; 2013;2013:478516. (PMID: 24063005)
- Hirayama K. Nonprogressive juvenile spinal muscular atrophy of the distal upper limb. In: De Jong JM, editor. Handbook of Clinical Neurology. Amsterdam, Netherlands: Elsevier; 1991. pp. 107–20. [Google Scholar]

